


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
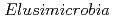 are metabolically diverse compared to gut microbiome and some have a novel nitrogenase paralog
are metabolically diverse compared to gut microbiome and some have a novel nitrogenase paralogM heust, R.*; Castelle, C. J.*; Matheus Carnevali, P. B.*; Farag, I. F.*; He, C.*; Chen, L.-X.*; 天野 由記; Hug, L. A.*; Banfield, J. F.*
heust, R.*; Castelle, C. J.*; Matheus Carnevali, P. B.*; Farag, I. F.*; He, C.*; Chen, L.-X.*; 天野 由記; Hug, L. A.*; Banfield, J. F.*
ISME Journal, 14(12), p.2907 - 2922, 2020/12
被引用回数:40 パーセンタイル:94.4(Ecology)We reconstructed 94 draft-quality, non-redundant genomes from diverse animal-associated and natural environments. Genomes group into 12 clades, 10 of which previously lacked reference genomes. Groundwater-associated are predicted to be capable of heterotrophic or autotrophic lifestyles, reliant on oxygen or nitrate/nitrite-dependent respiration of fatty acids, or a variety of organic compounds and Rnf-dependent acetogenesis with hydrogen and carbon dioxide as the substrates. Genomes from two clades of groundwater-associated often encode a new homologous group of nitrogenase-like proteins that co-occur with an extensive suite of radical SAM-based proteins. We identified similar genomic loci in genomes of bacteria from the Gracilibacteria and Myxococcus phyla and predict that the gene clusters reduce a tetrapyrrole, possibly to form a novel cofactor. The animal-associated clades nest phylogenetically within two groundwater-associated clades. Thus, we propose an evolutionary trajectory in which some adapted to animal-associated lifestyles from groundwater-associated species via genome reduction.
Al-Shayeb, B.*; Sachdeva, R.*; Chen, L.-X.*; Ward, F.*; Munk, P.*; Devoto, A.*; Castelle, C. J.*; Olm, M. R.*; Bouma-Gregson, K.*; 天野 由記; et al.
Nature, 578(7795), p.425 - 431, 2020/02
被引用回数:223 パーセンタイル:99.47(Multidisciplinary Sciences)Phage typically have small genomes and depend on their bacterial hosts for replication. We generated metagenomic datasets from many diverse ecosystems and reconstructed hundreds of huge phage genomes, between 200 kbp and 716 kbp in length. Thirty four genomes were manually curated to completion, including the largest phage genomes yet reported. Expanded genetic repertoires include diverse and new CRISPR-Cas systems, tRNAs, tRNA synthetases, tRNA modification enzymes, initiation and elongation factors and ribosomal proteins. Phage CRISPR have the capacity to silence host transcription factors and translational genes, potentially as part of a larger interaction network that intercepts translation to redirect biosynthesis to phage-encoded functions. Some phage repurpose bacterial systems for phage-defense to eliminate competing phage. We phylogenetically define seven major clades of huge phage from human and other animal microbiomes, oceans, lakes, sediments, soils and the built environment. We conclude that large gene inventories reflect a conserved biological strategy, observed across a broad bacterial host range and resulting in the distribution of huge phage across Earth's ecosystems.
